
Anastrozole in Men on TRT: What It Does, When It Helps, and When It Doesn’t
Many men on testosterone therapy are prescribed anastrozole as a matter of routine. But blanket estrogen suppression often creates the very problems it’s meant to fix—including, paradoxically, the gynecomastia it was supposed to prevent.
You started testosterone therapy hoping to feel sharper, stronger, and more like yourself. But a few weeks in, your doctor added another prescription to the mix—anastrozole—with a brief explanation about estrogen. Maybe your levels came back elevated. Maybe it was just standard protocol.
If you’ve ever wondered what anastrozole is actually doing, whether you need it, and what happens when it’s used too aggressively, this post is for you. The answers might surprise you—especially the part about gynecomastia.
Why Estrogen Matters in Men
Estrogen is not a female hormone. It’s a steroid hormone that men produce, require, and depend on for basic physiological function. In men, the primary form is estradiol (E2), produced largely through a conversion process called aromatization—testosterone is converted to estradiol by an enzyme called aromatase. More than 80% of circulating estradiol in men comes from this peripheral conversion.
Estradiol does a remarkable amount of work in the male body. It plays a central role in bone density, cardiovascular health, lipid regulation, cognition, mood, sleep quality, and libido. Estradiol is now understood to be the dominant regulator of bone resorption in men—not testosterone. Men with very low estradiol develop osteoporosis. Men with suppressed estradiol on TRT often report joint pain, low libido, depression, and brain fog—the same symptoms they were trying to fix with testosterone in the first place.
The problem isn’t estrogen itself. It’s estradiol that falls too low when aromatase is over-suppressed, or rises too high relative to testosterone. That balance—not any single number in isolation—is what matters clinically.
Estradiol isn’t the enemy on TRT. The relationship between testosterone and estradiol matters far more than either number read in isolation.
What Anastrozole Actually Does
Anastrozole is an aromatase inhibitor (AI). It blocks aromatase, the enzyme responsible for converting testosterone to estradiol. When aromatase activity is suppressed, less testosterone converts—and circulating estradiol drops.
It is FDA-approved for breast cancer treatment and prevention in postmenopausal women. Its use in men on TRT is entirely off-label, meaning it’s prescribed based on clinical reasoning rather than a specific indication in the drug’s labeling. That doesn’t make it inappropriate—off-label prescribing is standard in hormone medicine—but it does mean the prescriber bears responsibility for the clinical judgment behind each prescription.
Anastrozole is typically prescribed as a low-dose oral tablet in the TRT context, often 0.25 mg to 0.5 mg taken two to three times per week. The goal isn’t to eliminate estradiol. The goal is to keep testosterone and estradiol in a healthy relationship to each other—and that relationship looks different depending on where your testosterone level is optimized.
The Evidence on Estradiol and Sexual Function
One of the most important studies in this area was published in the New England Journal of Medicine in 2013. Finkelstein and colleagues enrolled 400 healthy men, suppressed their endogenous testosterone and estradiol, then randomized them to receive varying doses of testosterone—with or without an aromatase inhibitor to block estradiol conversion.
When estradiol was suppressed alongside testosterone, sexual function declined significantly—even in men who had adequate testosterone levels. Sexual desire and erectile function tracked with estradiol, not with testosterone alone. The conclusion was direct: estradiol plays an independent and essential role in male sexual function. A separate retrospective study further found that the estradiol-to-testosterone ratio independently predicted erectile function after controlling for other variables—reinforcing that the balance between these two hormones matters more than either value in isolation.
This matters directly for how anastrozole is used in practice. If an aromatase inhibitor is suppressing estradiol below the functional range, sexual function often declines—not improves. The drug intended to optimize TRT may be neutralizing its most meaningful benefit.
Why a Fixed Estradiol Target Misses the Point
For years, many TRT protocols aimed to keep estradiol within a fixed absolute range—often somewhere in the 20–40 pg/mL band. That target was derived from population reference ranges built around average men with average testosterone levels. It doesn’t hold when testosterone is optimized significantly higher.
The more clinically meaningful tool is the testosterone-to-estradiol ratio, or T:E2, calculated by dividing total testosterone in ng/dL by estradiol in pg/mL. Importantly, this ratio has two meaningful edges—a floor and a ceiling—and clinical problems arise at either end. A 2025 comprehensive review examining T:E2 data across multiple large population studies described a beneficial range of approximately 10 to 30. Below 10, estradiol is high relative to testosterone and estrogen-dominant symptoms—water retention, gynecomastia, reduced libido—become more likely. Above 30, estradiol is insufficient relative to testosterone and the low-estradiol picture emerges: joint pain, brain fog, poor sleep, reduced bone protection, and impaired sexual function.
A study in men with hypogonadotropic hypogonadism established a T:E2 ratio of 12 as a clinically meaningful lower threshold, below which erectile function was significantly impaired. But the lower bound only tells part of the story. A man with testosterone of 1,000 ng/dL and estradiol of 1 pg/mL has a T:E2 of 1,000—technically above that threshold, but clearly estradiol-deficient and symptomatic. The ratio only functions as a useful clinical tool when both edges of the range are respected.
Consider the math in an optimized TRT context: if testosterone is 2,000 ng/dL and estradiol is 20 pg/mL, the T:E2 ratio is 100—well above the upper bound of 30. That isn’t controlled estrogen; it’s estrogen deficiency in a high-androgen environment. The same estradiol of 20 pg/mL in a man with testosterone of 400 ng/dL produces a T:E2 of 20, which falls comfortably within the healthy range. Same estradiol number, opposite clinical meaning. This is why fixed absolute targets fail men on optimized TRT.
The T:E2 ratio has a floor and a ceiling. Most over-suppression errors on TRT are violations of the upper bound—not the lower one.
The Gynecomastia Paradox: How Suppressing Estrogen Can Cause What You’re Trying to Prevent
One of the most common reasons anastrozole is added to a TRT protocol is fear of gynecomastia—the development of glandular breast tissue in men. That concern is clinically legitimate. When estradiol rises significantly relative to testosterone, it can stimulate ductal proliferation in breast tissue, and no man starting TRT wants that outcome.
The clinical irony is that overly aggressive aromatase inhibition can promote both forms of breast enlargement men want to avoid—through a two-step pathway that runs through insulin resistance, ectopic fat deposition, and local aromatase activity in breast tissue.
Step One: Anastrozole, Insulin Resistance, and Ectopic Fat Redistribution
A double-blind randomized crossover study in healthy men found that six weeks of anastrozole therapy produced a statistically significant reduction in insulin sensitivity, measured by hyperinsulinemic-euglycemic clamp. The authors concluded that local estrogen production in skeletal muscle plays an important role in peripheral insulin sensitivity, and that aromatase inhibition disrupts this. The same study noted a reduction in leptin and raised the possibility of a shift from subcutaneous toward visceral fat deposition.
This is consistent with the Finkelstein 2013 New England Journal of Medicine data showing that estradiol—not testosterone—is the primary regulator of fat mass in men. When estradiol was suppressed in that trial, fat accumulation increased regardless of testosterone level.
When insulin resistance drives fat redistribution toward visceral and ectopic depots—including the chest wall and breast region—the result is pseudogynecomastia: breast enlargement from fat deposition rather than glandular proliferation. This is clinically distinct from true gynecomastia, produces the same unwanted cosmetic outcome, and does not respond to further anastrozole use.
Step Two: Ectopic Breast Fat as a Local Aromatase Source
Breast adipose tissue is not metabolically inert. It contains active aromatase and converts androgens to estrogens locally, generating an estrogen-rich microenvironment within breast tissue itself—independent of what circulating estradiol levels show on a blood draw.
The clinical literature on gynecomastia in obese men documents this pathway explicitly. Increased aromatase activity in adipose tissue, combined with leptin’s capacity to stimulate aromatase in breast tissue and directly promote epithelial cell growth, is recognized as a contributor to true glandular gynecomastia in overweight and obese men—not just pseudogynecomastia.
So the paradox closes: anastrozole prescribed to prevent gynecomastia promotes insulin resistance and ectopic fat redistribution toward the breast region—producing pseudogynecomastia—and that accumulated breast adipose tissue then becomes a local estradiol source capable of driving true glandular proliferation. The AI blocks circulating aromatase while inadvertently creating a new local aromatase depot in exactly the tissue it was meant to protect.
To be clear about the evidence: each mechanistic link in this sequence is supported by peer-reviewed data. The complete pathway from anastrozole use to glandular gynecomastia has not been studied as a direct clinical endpoint in TRT patients specifically. The inference is mechanistically coherent and grounded in the underlying biology—but should be understood as an evidence-informed argument rather than a proven causal chain in the TRT population.
The drug prescribed to prevent gynecomastia can promote the fat redistribution and local aromatase activity that drive both forms of breast enlargement it was meant to stop.
When Anastrozole Is Clinically Justified
There are men on TRT who genuinely benefit from anastrozole. This happens when estradiol rises high enough relative to testosterone to produce clear, confirmed symptoms—and when labs support that picture.
Symptoms That May Indicate Elevated Estradiol
Nipple tenderness or early glandular development is one of the more objective early signs that estradiol has risen significantly relative to testosterone. Gynecomastia that progresses to established glandular tissue can be permanent even after estradiol is corrected, so acting early matters. Water retention around the midsection and face, emotional lability, and difficulty with erections despite adequate testosterone levels are other symptoms that—in the context of a low T:E2 ratio on labs—constitute a reasonable clinical case for considering an AI.
What the Labs Should Show
Symptoms alone aren’t enough to justify starting an aromatase inhibitor. Estradiol should be measured using a sensitive assay—not a standard immunoassay, which is less accurate in men at lower estradiol ranges. Total and free testosterone should be drawn at the same time so the T:E2 ratio can be calculated in context. When the ratio falls below 10 in the presence of symptomatic estrogen excess, a clinical conversation about anastrozole is reasonable. Without both—symptoms and a ratio that warrants intervention—treating a number on a page is not good medicine.
When Anastrozole Does More Harm Than Good
Anastrozole is routinely co-prescribed with TRT in many practices as a preventive measure, regardless of symptoms or ratio. The reasoning is intuitive—more testosterone means more aromatization, so add an AI to stay ahead of it. In practice, this approach suppresses estradiol in men who didn’t need suppression in the first place, and sets in motion the metabolic cascade described above.
The symptoms of iatrogenic low estradiol look remarkably like the symptoms of low testosterone: joint pain and stiffness, low libido, poor sleep, mood changes, fatigue, and brain fog. Add the long-term risk of reduced bone density from sustained suppression, and the risk-benefit calculation for reflexive AI prescribing becomes difficult to justify.
The High Converter Who Doesn’t Need an AI
Some men convert testosterone to estradiol at higher rates due to body composition, genetics, and age. A man with higher adiposity may show estradiol well above typical reference ranges on TRT and feel completely well—because his T:E2 ratio is intact given his testosterone level. If he has no symptoms of estrogen excess and his ratio is in range, prescribing an aromatase inhibitor is intervening where no intervention was needed—and risks triggering the insulin resistance and fat redistribution cycle described above.
The more appropriate target in that scenario is body composition. Reducing adipose tissue reduces systemic aromatase activity and improves the ratio without pharmacologic suppression.
The Sensitivity Problem
Anastrozole’s effect on estradiol is not perfectly predictable. Individual responses vary significantly—some patients take a small dose and crash their estradiol, while others require higher doses for the same effect. Fixed-dose protocols prescribed reflexively carry real risk of producing iatrogenic estrogen deficiency that may take weeks to resolve and that looks, on labs and symptoms, indistinguishable from the problem you were trying to treat.
A Pattern Worth Recognizing
A patient came in having been on TRT for about eighteen months. Testosterone levels were well above 1,000 ng/dL, but the patient continued to report fatigue, joint stiffness, and a complete absence of the libido improvement that had initially prompted therapy. The protocol included anastrozole at 0.5 mg three times per week.
Estradiol came back at 8 pg/mL. T:E2 ratio: above 125—well beyond the upper bound of the healthy range. That’s not estrogen management; that’s estrogen deficiency created by the treatment itself. After discontinuing anastrozole and allowing estradiol to recover over several weeks, most of the persistent symptoms resolved. Testosterone hadn’t changed. What changed was giving estradiol room to do its job.
This pattern is not unusual. It’s the reason experienced hormone clinicians approach aromatase inhibitors with a strong preference for evidence-first prescribing: symptoms plus ratio-contextualized labs, not reflex prophylaxis.
Monitoring If You Are on Anastrozole
If anastrozole is part of your current protocol, monitoring should include sensitive estradiol and total and free testosterone drawn together so the T:E2 ratio can be tracked at each timepoint. Every three months while titrating; longer intervals once stable.
If you’re experiencing joint pain, low libido, mood changes, or fatigue on a protocol that includes an AI, low estradiol should be on the differential before assuming the testosterone dose needs to go up. Bone density is worth monitoring beyond one year of use, given estradiol’s dominant role in male skeletal maintenance.
What to Do If You’re Not Sure Your Protocol Is Right
If you’re on TRT and not feeling the way you expected to—especially if your protocol includes an aromatase inhibitor—a thorough review of your labs in the context of your symptoms is a reasonable first step. The right question isn’t whether your testosterone number is high enough. It’s whether your testosterone and estradiol are in the right relationship to each other, and whether what you’re taking is actually working for you or against you.
At Precision Hormone Consulting, this is exactly the kind of case we see regularly: patients who are technically on therapy but not benefiting from it, often because one variable in the protocol is undermining everything else. Getting it right requires time, attention, and a willingness to look at the full picture rather than manage individual numbers in isolation.
We offer a free initial consultation, available virtually throughout Texas and Arizona, or by calling the clinic at 832-281-5199. It’s a conversation, not a commitment—and it costs you nothing to find out whether something in your current plan deserves a closer look.
References
1. Finkelstein JS, Lee H, Burnett-Bowie SA, Pallais JC, Yu EW, Borges LF, et al. Gonadal steroids and body composition, strength, and sexual function in men. N Engl J Med. 2013;369(11):1011–1022. doi: 10.1056/NEJMoa1206168. PMID: 24024838.
2. Khosla S, Melton LJ 3rd, Riggs BL. Clinical review 144: estrogen and the male skeleton. J Clin Endocrinol Metab. 2002;87(4):1443–1450. PMID: 11932262.
3. Pan B, Ye H, Huang Y, et al. Relationship between penile erection and the ratio of estradiol to testosterone: A retrospective study. Andrologia. 2020;52(9):e13701. PMID: 32167186.
4. Seaman E, Bernstein A, Bhindi R, et al. A review on testosterone: estradiol ratio—does it matter, how do you measure it, and can you optimize it? World J Mens Health. 2025;43(3):458–471. doi: 10.5534/wjmh.240029.
5. Aydın A, Selvi I, Basar H. Role of testosterone to estradiol ratio in predicting the efficacy of recombinant human chorionic gonadotropin and testosterone treatment in male hypogonadism. Int Urol Nephrol. 2023;55(10):2603–2609. PMID: 37634764.
6. Gibb FW, Homer NZM, Faqehi AMM, et al. Aromatase inhibition reduces insulin sensitivity in healthy men. J Clin Endocrinol Metab. 2016;101(5):2040–2048. doi: 10.1210/jc.2015-4146. PMID: 26990060.
7. Grossmann M, Cheung AS, Lasaitiene D, et al. Adipose tissue dysfunction and obesity-related male hypogonadism. Clin Endocrinol (Oxf). 2022;97(1):11–21. PMID: 35446432.
8. Braunstein GD. Gynecomastia: etiology, diagnosis, and treatment. In: Feingold KR, et al., eds. Endotext. MDText.com; 2023. Available at: https://www.ncbi.nlm.nih.gov/books/NBK279105/
9. Johnson RE, Murad MH. Gynecomastia: pathophysiology, evaluation, and management. Mayo Clin Proc. 2009;84(11):1010–1015. PMID: 19880690.
Medical Disclaimer: This content is for educational purposes only and does not constitute medical advice, diagnosis, or treatment. Anastrozole is an FDA-approved medication used off-label in the context of testosterone replacement therapy. Individual clinical decisions should be made in consultation with a licensed physician who has reviewed your complete medical history and current laboratory values. Do not adjust or discontinue any prescribed medication without guidance from your treating provider.
Visceral Fat Is a Hormone Problem — Not a Willpower Problem
If you're doing everything right and still carrying stubborn abdominal fat, the problem may not be discipline — it may be your hormones. Learn why visceral fat is a metabolic and hormonal problem, and how we assess and treat it at Precision Hormone Consulting.
If you've cleaned up your diet, started lifting weights, cut back on alcohol, and you're still carrying stubborn fat around your midsection — the frustration is understandable. Most people in that position assume they need to do more, or try harder. What they actually need is better information.
Abdominal fat is not simply the result of caloric excess or insufficient discipline. For a significant portion of people — particularly those in midlife — stubborn visceral fat is a hormonal and metabolic problem. It responds to hormonal and metabolic solutions. Understanding why requires a closer look at what visceral fat actually is.
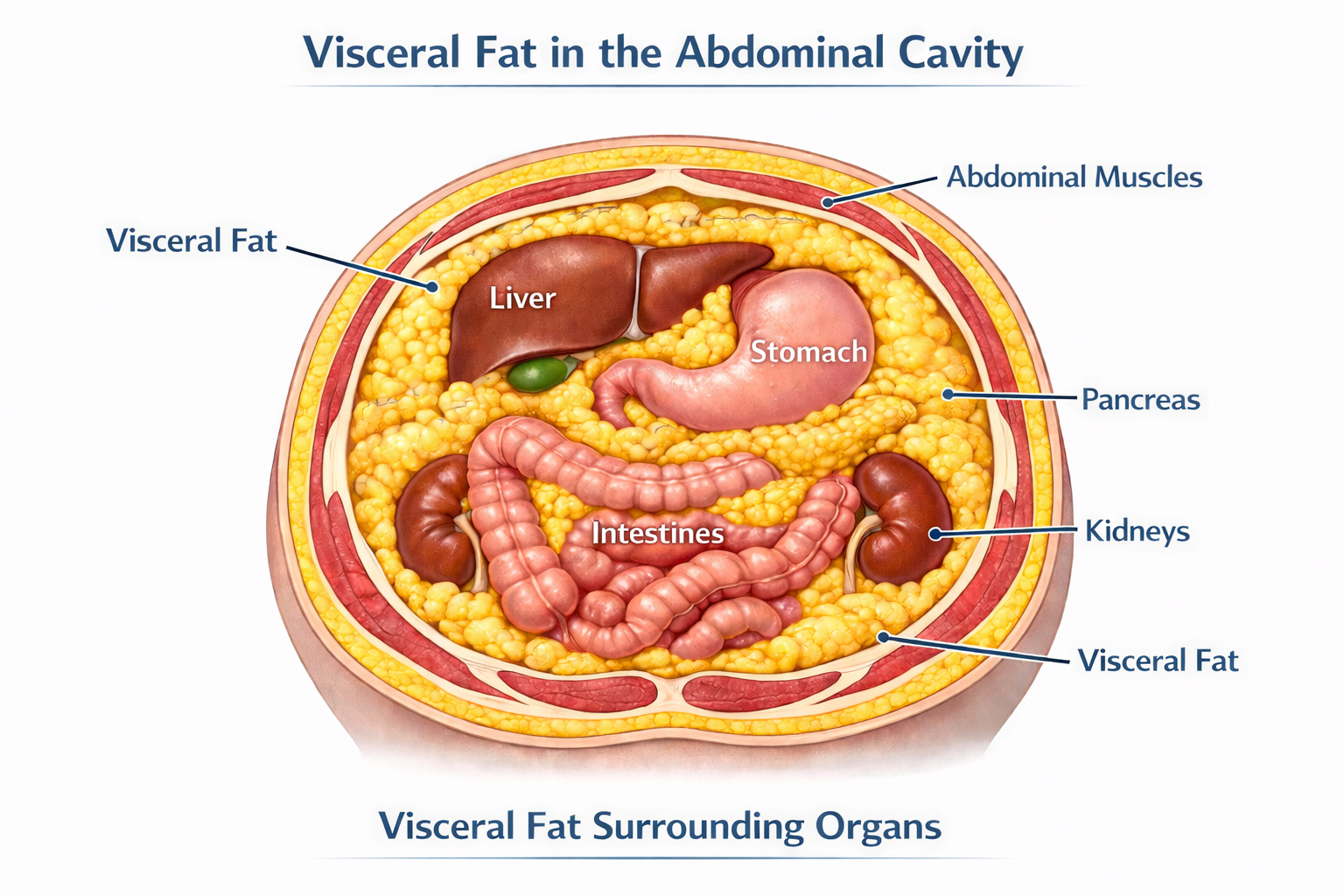
Not All Body Fat Behaves the Same Way
The fat you can pinch just beneath the skin is called subcutaneous fat. It's not metabolically inert, but it's relatively benign compared to the fat that accumulates deep inside the abdominal cavity — surrounding the liver, pancreas, and intestines. That's visceral fat, and it behaves very differently.
Visceral adipose tissue is biologically active. It functions, in many respects, as an endocrine organ — secreting hormones, driving inflammation, and interfering with the metabolic systems that govern energy, appetite, and hormonal balance. The technical term for these secreted compounds is adipokines, and their effects ripple throughout the body.
When visceral fat is elevated, leptin — the hormone that signals satiety — becomes dysregulated, contributing to persistent hunger even in the context of adequate intake. Adiponectin, a hormone that improves insulin sensitivity and reduces inflammation, declines. Pro-inflammatory cytokines like TNF-α and IL-6 rise, creating a state of chronic low-grade systemic inflammation that quietly drives cardiovascular risk, metabolic dysfunction, and hormonal disruption.
Visceral fat is also rich in aromatase — an enzyme that converts androgens like testosterone into estrogens. This is not a minor detail. It means that excess visceral fat doesn't just accumulate as a downstream effect of hormonal imbalance; it actively worsens that imbalance, accelerating androgen breakdown and feeding back on the very hormonal systems that keep fat distribution and metabolism healthy.
Visceral fat doesn't just respond to hormone imbalance — it creates it.
The Hormones Driving Visceral Fat Accumulation
Visceral fat and hormonal dysfunction reinforce each other. Addressing one without the other is rarely sufficient. Here's what the evidence shows for each major hormone axis:
Testosterone
Low testosterone is both a cause and a consequence of visceral fat accumulation. Testosterone promotes lean muscle mass and healthy fat distribution. As levels decline — which happens gradually in both men and women with age — fat preferentially shifts toward the abdomen. That visceral fat then accelerates testosterone breakdown through aromatization, producing more estrogen and suppressing the signaling pathway between the brain and the gonads. The result is a self-reinforcing cycle that worsens over time without intervention.
Estradiol
In women, estradiol plays an underappreciated role in metabolic health. It promotes favorable fat distribution, supports insulin sensitivity, and reduces systemic inflammation. The decline of estradiol at perimenopause and menopause is one of the primary drivers of the visceral fat accumulation many women notice in their 40s and 50s — even without meaningful changes in diet or activity level. Restoring physiologic estradiol is a legitimate metabolic intervention, not merely a quality-of-life measure.
DHEA
DHEA is the most abundant circulating steroid hormone in the body, and it declines significantly with age. Low DHEA correlates with increased visceral adiposity, reduced insulin sensitivity, and an elevated inflammatory state. It receives less attention than testosterone or estradiol in mainstream medicine, but it's a meaningful part of a comprehensive hormonal assessment.
Thyroid — Specifically Free T3
Thyroid hormone drives thermogenesis, fat oxidation, and insulin sensitivity. The clinically relevant form is Free T3 — the metabolically active fraction. Many standard workups stop at TSH, missing patients whose Free T3 is suboptimal even when TSH appears normal. When Free T3 is low, the metabolic engine slows: fat oxidation decreases, insulin resistance worsens, and visceral fat accumulates even in patients doing everything else right.
Cortisol
Visceral adipocytes have a high concentration of glucocorticoid receptors, making them exquisitely responsive to cortisol — the body's primary stress hormone. Chronic psychological or physiological stress translates directly into central fat accumulation, elevated blood glucose, and worsening insulin resistance. Cortisol dysregulation isn't optional to address in any serious approach to metabolic health.
Insulin Resistance
Insulin resistance and visceral fat are so tightly intertwined that separating cause from consequence is often impossible. Visceral fat drives insulin resistance through adipokine dysregulation, chronic inflammation, and excess free fatty acid release into the portal circulation. Insulin resistance, in turn, creates a hormonal environment that favors further visceral fat accumulation. Addressing one without the other rarely produces durable results.
What We Measure — and Why It Matters
Identifying visceral fat burden and its downstream metabolic effects requires looking beyond a scale or a BMI table. BMI, in particular, tells you almost nothing about where fat is distributed or how metabolically active it is. Two people with identical BMIs can carry dramatically different metabolic risk.
At Precision Hormone Consulting, a comprehensive assessment includes:
Waist circumference and waist-to-hip ratio are simple but meaningful starting points — far more predictive of metabolic risk than weight or BMI alone. In-office, we use InBody bioelectrical impedance analysis to go further, generating a validated estimate of visceral fat, lean mass, and body composition that gives us a quantitative baseline and a way to track changes over time. The clinical gold standards for visceral fat measurement — DEXA and MRI — offer greater precision but are expensive and largely inaccessible outside of research settings. For the purposes of clinical monitoring, our combination of anthropometric measures and InBody analysis provides a practical, actionable picture of visceral fat burden without requiring a radiology referral.
Fasting insulin and HOMA-IR — the most direct available measures of insulin resistance. A fasting glucose in the normal range can mask significantly elevated insulin levels, which is where the metabolic damage is already occurring.
Triglyceride/HDL ratio — an accessible and underutilized surrogate for insulin resistance and small, dense LDL particle burden. A standard lipid panel showing normal total cholesterol can coexist with substantial cardiovascular risk in a patient with visceral adiposity and insulin resistance.
hs-CRP — high-sensitivity C-reactive protein, used as a marker of systemic low-grade inflammation driven by visceral fat.
Adiponectin — an inverse marker of visceral fat and insulin resistance. Low levels indicate significant metabolic risk even before glucose dysregulation becomes overt on a standard chemistry panel.
SHBG (Sex Hormone Binding Globulin) — low SHBG is a reliable early signal of hepatic insulin resistance, often appearing before other markers become abnormal. It is particularly useful in women as an early warning sign of metabolic dysfunction.
LH/FSH ratio — in reproductive-age women, normal physiology produces an FSH level approximately twice that of LH. When insulin resistance is present, this ratio begins to narrow — sometimes approaching 1:1 — even before other metabolic markers are overtly abnormal. This is an early, underutilized signal of insulin's effect on the hormonal axis, and it does not require a PCOS diagnosis to be clinically meaningful.
These markers, taken together, provide a far more complete picture of metabolic health than any single value.
The PHC Approach: Treating the Root Cause
A patient came to us in their mid-forties — lean by BMI standards, active, eating well. Their complaint was persistent abdominal fullness and fatigue that had been gradually worsening for two years. Standard labs from their primary care physician had come back normal. Our panel told a different story: suboptimal Free T3, low SHBG, an elevated fasting insulin consistent with early insulin resistance, and a testosterone level that was technically within the reference range but well below what we'd expect for their age and activity level. Within four months of a targeted protocol, their body composition had shifted meaningfully and their energy had returned.
That kind of presentation is common. The tools to identify and address it are available — they just aren't part of routine care.
Hormone Optimization
Restoring physiologic hormone levels is one of the most effective metabolic interventions available. Testosterone optimization in both men and women improves lean muscle mass, reduces visceral fat, and enhances insulin signaling. Estradiol replacement — particularly relevant around perimenopause and menopause — shifts fat distribution favorably and supports metabolic function. DHEA optimization reduces inflammation and supports body composition. Ensuring Free T3 is in an optimal range, not merely a "not flagged" range, restores the metabolic rate that drives fat oxidation.
Peptide Therapy
Growth hormone-releasing peptides are a valuable adjunct for patients with significant visceral fat burden. Tesamorelin has demonstrated specific efficacy in visceral fat reduction in clinical trials. CJC-1295/Ipamorelin combinations support broader growth hormone axis optimization, improving body composition, sleep quality, and recovery.
GLP-1 Medications
For patients with significant metabolic burden or insulin resistance, GLP-1 receptor agonists represent one of the most effective pharmacologic tools currently available. Their mechanisms go well beyond appetite suppression — they improve insulin signaling, reduce hepatic fat, lower systemic inflammation, and produce meaningful, sustained reductions in visceral adiposity.
It's worth noting that GLP-1 is itself a peptide hormone produced naturally in the gut. In patients with metabolic dysfunction, endogenous GLP-1 production is often impaired — meaning these medications are, in a meaningful sense, optimizing a hormone that the body is no longer producing adequately. That framing fits squarely within a hormone optimization model rather than a weight loss drug model.
Lifestyle Integration
No clinical protocol works in isolation. Resistance training, protein-adequate nutrition, quality sleep, and deliberate stress management all independently reduce visceral adiposity and improve insulin sensitivity. Our role is to help patients optimize the full picture — not simply prescribe and monitor. Clinical intervention amplifies the results of good lifestyle fundamentals; it doesn't replace them.
If You've Been Doing the Right Things and Still Not Getting Results
The frustration of doing everything by the book and still carrying stubborn abdominal fat is real — and it usually means something in the hormonal or metabolic picture hasn't been identified yet.
Visceral fat is not a character flaw. It is a metabolic and hormonal problem, and it responds to metabolic and hormonal solutions. The evidence is clear: optimizing testosterone, estradiol, DHEA, thyroid, and metabolic markers produces real, measurable improvements in body composition and long-term health outcomes.
At Precision Hormone Consulting, we specialize in exactly this kind of comprehensive, root-cause evaluation. If you're ready to understand what's actually driving your metabolic health — and address it systematically — we'd be glad to have that conversation.
Schedule a free consultation at precisionhormoneconsulting.com, or call the clinic to book an appointment. Virtual and in-person options are both available.
[DISCLAIMER] This content is for educational purposes only and does not constitute medical advice. Hormone optimization, peptide therapy, and GLP-1 medications involve prescription therapies that require individualized evaluation, monitoring, and ongoing clinical oversight. Some therapies discussed may be used off-label. Results vary. Consult a qualified physician before beginning any new treatment protocol.